
A successful regulatory submission to the FDA begins long before a sponsor uploads documents through the Electronic Submissions Gateway.
Many delays are not caused by weak science, but by early gaps in regulatory strategy, incomplete submission documents, inconsistent clinical data, poor eCTD planning, or delayed cross-functional coordination.
For pharma, biotech, medical device, and CRO teams, regulatory submission cannot be just an online filing task. It has to be the outcome of strategy, medical writing, clinical data management, quality review, publishing, and regulatory operations working together.
This guide explains the FDA submission process step by step while highlighting where expert regulatory submission services can add value.
TL; DR:
The FDA regulatory submission process involves identifying the correct submission pathway, preparing compliant regulatory submission documents, organizing them in eCTD format, validating the submission package, submitting electronically through the FDA Electronic Submissions Gateway (ESG), and managing FDA review queries until approval or clearance.
The process sounds straightforward, but every step requires precision. Even minor errors can trigger review delays, additional information requests, or refusal-to-file notices.
What Is a Regulatory Submission to the FDA?
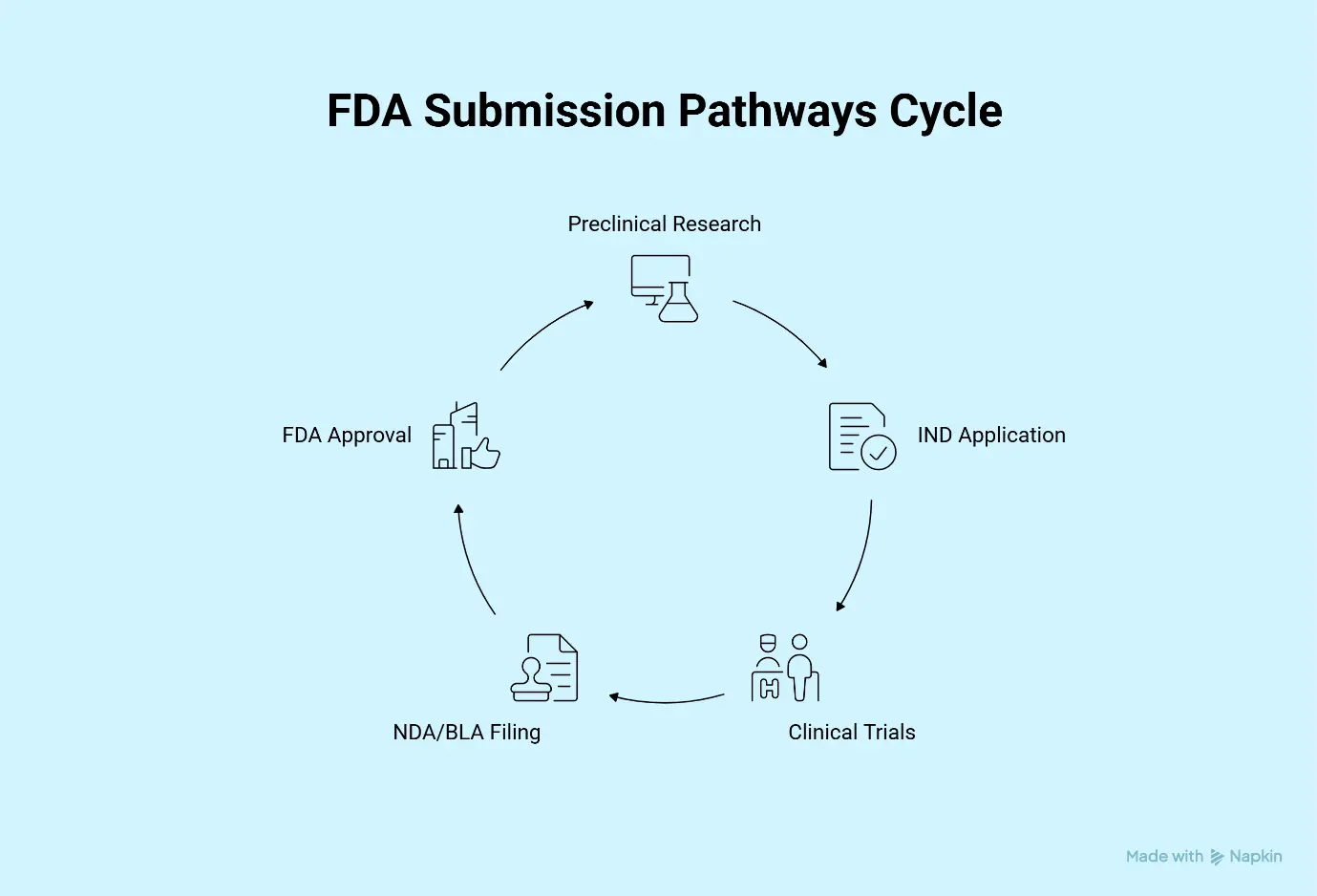
A regulatory submission is the formal package of scientific, clinical, manufacturing, and administrative information submitted to the FDA to obtain authorization for clinical trials, product approval, or market clearance.
Depending on the product type, submissions may include:
- Investigational New Drug (IND) applications
- New Drug Applications (NDA)
- Biologics License Applications (BLA)
- Abbreviated New Drug Applications (ANDA)
- Investigational Device Exemptions (IDE)
- 510(k) submissions
- Premarket Approval (PMA) applications
The FDA reviews these submissions to determine whether a product is safe, effective, and manufactured according to applicable standards.
Which FDA Submission Pathway Should You Choose?
The correct submission pathway depends on whether your product is a drug, biologic, generic medicine, or medical device. Selecting the wrong pathway can create unnecessary delays and additional regulatory questions.
| Submission Type | Purpose |
| IND (Investigational New Drug Application) | Authorization to begin clinical trials |
| NDA (New Drug Application) | Approval for a new drug |
| BLA (Biologics License Application) | Approval for biologics |
| ANDA (Abbreviated New Drug Application) | Approval for generic drugs |
| IDE (Investigational Device Exemption) | Device clinical investigations |
| 510(k) | Device market clearance |
| PMA (Premarket Approval) | High-risk device approval |
Before compiling regulatory submission documents, sponsors should evaluate product classification, intended use, risk profile, and applicable regulatory requirements.
What Regulatory Submission Documents Does the FDA Require?
The specific requirements vary by submission type, but most submissions include the following components:
Administrative Documentation
- Cover letters
- FDA forms
- Financial disclosures
- Certifications
Quality and Manufacturing Documentation
- Chemistry, Manufacturing, and Controls (CMC)
- Stability studies
- Validation reports
- Manufacturing process descriptions
Nonclinical Documentation
- Toxicology studies
- Pharmacology reports
- Animal study data
Clinical Documentation
- Clinical protocols
- Clinical study reports
- Statistical analysis plans
- Safety summaries
Labelling and Product Information
- Prescribing information
- Product labels
- Risk management documentation
Incomplete or inconsistent regulatory submission documents are among the most common reasons for regulatory delays.
How Should Regulatory Documents Be Organized in eCTD Format?
Most FDA drug and biologic submissions must be organized using the Electronic Common Technical Document (eCTD), the global standard for electronic regulatory submissions.
The eCTD structure contains 5 modules:
| eCTD Module | Contents |
| Module 1 | Administrative information |
| Module 2 | Summaries and overviews |
| Module 3 | Quality and CMC (Chemistry, Manufacturing, and Controls) data |
| Module 4 | Nonclinical reports |
| Module 5 | Clinical study reports |
The FDA requires sponsors to submit electronic dossiers in a structured format that enables efficient review and lifecycle management. Proper eCTD organization reduces review complexity and supports global regulatory submissions across multiple health authorities.
How to Ensure Submission-Ready Documentation?
Many sponsors focus on compiling data but underestimate the importance of submission readiness.
Before submission, teams should verify:
- Data consistency across all documents
- Version control and document traceability
- Correct references and hyperlinks
- Statistical accuracy
- Compliance with submission specifications
- Medical writing quality
Industry experts strongly believe that regulatory success depends not only on the amount of data submitted but also on the quality, organization, and scientific integrity of that data.
How to Validate a Submission Before Filing?
Validation confirms that the submission meets technical requirements before it reaches FDA reviewers.
Common validation failures include:
- Missing files
- Broken hyperlinks
- Invalid XML structures
- Incorrect file naming conventions
- Corrupted PDFs
- Incomplete metadata
A technically compliant submission reduces the likelihood of avoidable review delays.
How to Submit Regulatory Documents Online to the FDA?
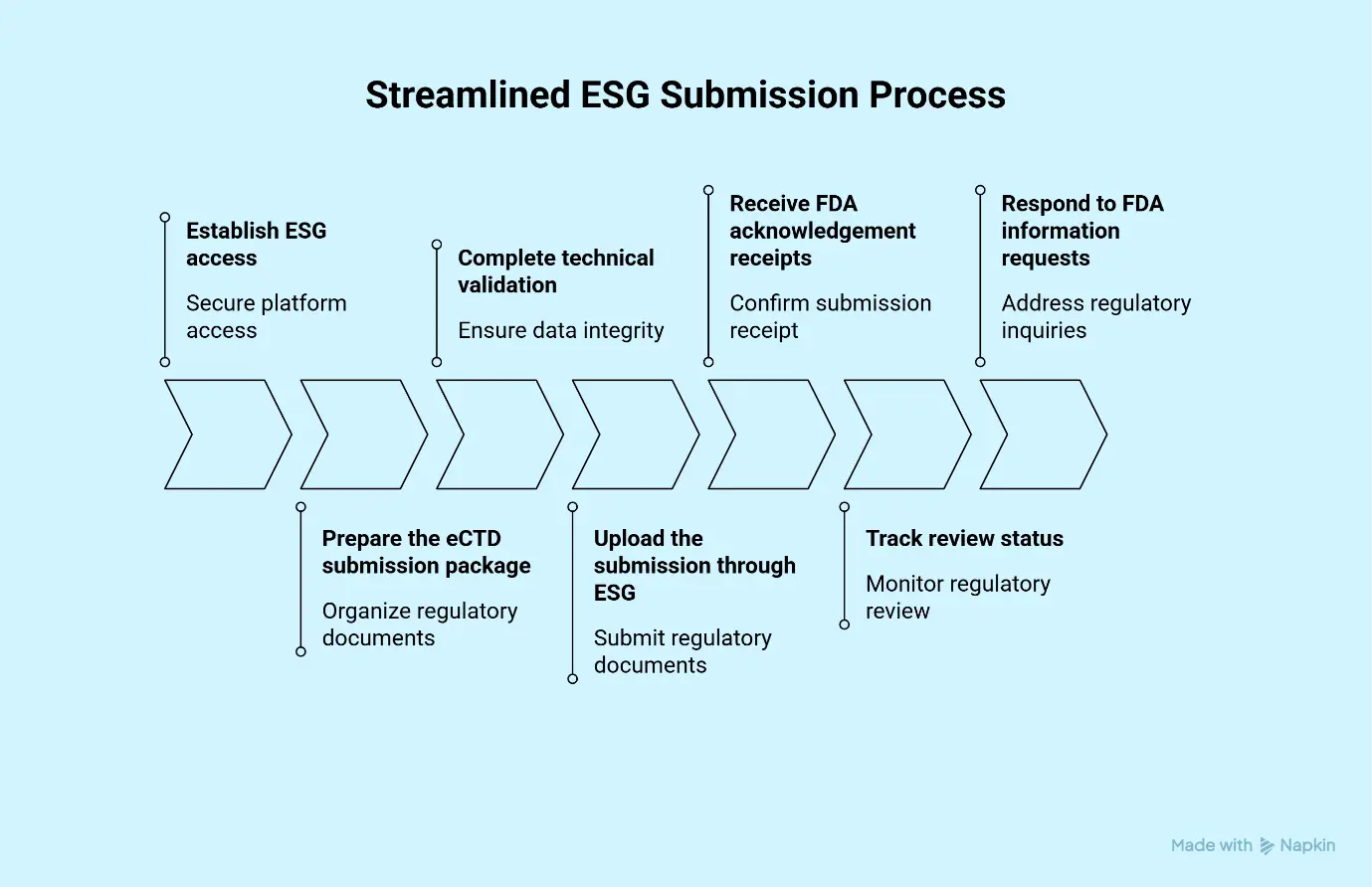
The FDA receives electronic submissions through the Electronic Submissions Gateway (ESG).
Step-by-Step Submission Process:
- Establish ESG access
- Prepare the eCTD submission package
- Complete technical validation
- Upload the submission through ESG
- Receive FDA acknowledgement receipts
- Track review status
- Respond to FDA information requests
The ESG serves as the FDA’s secure electronic submission platform and is a critical component of the modern regulatory affairs submission process.
What Happens After Submission?
Many organizations assume the process ends after filing.
In reality, submission management continues throughout the review cycle.
| Typical FDA Review Stage | Purpose |
| Technical Review | Verify submission integrity and compliance with electronic submission requirements |
| Filing Review | Assess application completeness and determine whether the submission is accepted for review |
| Scientific Review | Evaluate clinical, nonclinical, manufacturing, and safety data |
| Information Requests | Obtain clarifications, additional analyses, or supporting documentation from the sponsor |
| Final Decision | Issue approval, clearance, complete response, or rejection based on review findings |
The speed and quality of responses to FDA queries can significantly influence review timelines.
What Are the Most Common Reasons Regulatory Submissions Fail?
According to regulatory publishing experts, most failures stem from preventable issues rather than scientific shortcomings.
Common causes include:
- Incomplete regulatory submission documents
- Poor document version control
- Missing safety information
- Data inconsistencies
- eCTD formatting issues
- Failed technical validation
- Delayed responses to agency questions
Also read: Why Regulatory Submissions Fail: What Reviewers Expect in a High-Quality Regulatory Submission
What we at Innovate Research have seen is:
One of the most overlooked aspects of regulatory submission is cross-functional alignment. Regulatory teams, medical writers, biostatisticians, clinical operations teams, and data managers often work in parallel.
When these functions are not aligned early, inconsistencies can emerge during final dossier preparation, increasing the risk of review delays.
This aligns closely with Innovate Research’s integrated capabilities across regulatory affairs, medical writing, data management, biostatistics, and clinical operations.
How Do Regulatory Submission Services Improve Approval Readiness?
As submissions become increasingly complex, many sponsors rely on specialized regulatory submission services.
These services typically support:
- Regulatory strategy planning
- IND, NDA, BLA, ANDA, and IDE preparation
- Regulatory writing
- eCTD publishing
- Submission validation
- Regulatory operations
- Global regulatory submissions
- Agency interactions and responses
A strong regulatory partner helps ensure that submissions are scientifically sound, technically compliant, and aligned with agency expectations.
How Does Innovate Research Support Regulatory Submissions?
Innovate Research is a full-service CRO providing regulatory affairs and submission support for pharmaceutical, biotechnology, and medical device companies. Their services include:
- Regulatory strategy and intelligence
- IND preparation and submission
- NDA preparation and submission
- IDE preparation and submission
- eCTD publishing and submissions
- Regulatory writing
- Regulatory operations
- Regulatory agency liaison support
- Global regulatory submission support
The organization also integrates clinical operations, medical writing, data management, biostatistics, and quality assurance functions to support end-to-end regulatory readiness.
Also Read: How Innovate Research Simplifies Regulatory Submissions for Indian Pharma and Device Companies
FDA Submission Readiness Checklist
Before submission, confirm:
- Correct submission pathway selected
- Regulatory strategy finalized
- Regulatory submission documents complete
- Clinical and nonclinical data verified
- eCTD structure prepared
- Validation checks completed
- ESG access configured
- Internal quality review completed
- FDA response plan established
Final Thoughts
Submitting regulatory documents to the FDA online requires far more than uploading files. Success depends on selecting the right pathway, preparing accurate documentation, ensuring eCTD compliance, managing technical validation, and responding effectively throughout the review cycle.
For pharmaceutical, biotechnology, medical device, and CRO organizations, a well-planned regulatory submission strategy can reduce delays, improve review readiness, and accelerate the path to approval.
Innovate Research supports sponsors across every stage of the regulatory journey, from regulatory strategy and document preparation to eCTD publishing, submission management, and global regulatory submissions.
If your organization is preparing for an upcoming FDA filing, partner with Innovate Research. We ensure that your submission is complete, compliant, and approval-ready.
Frequently Asked Questions
1. What documents are required for FDA submission?
Most submissions require administrative forms, quality documentation, nonclinical reports, clinical data, safety information, and labelling materials.
2. What is eCTD?
eCTD is the electronic standard used to organize and submit regulatory information to the FDA and other global health authorities.
3. What is the FDA Electronic Submissions Gateway?
The ESG is the FDA’s secure platform for receiving electronic regulatory submissions.
4. How long does the FDA review process take?
Timelines vary depending on submission type, product complexity, review designation, and agency requests for additional information.
5. What are global regulatory submissions?
Global regulatory submissions involve preparing and adapting dossiers for multiple health authorities such as the FDA, EMA, MHRA, PMDA, and CDSCO.
6. When should companies use regulatory submission services?
Organizations often engage regulatory experts when preparing complex submissions, managing global filings, or operating with limited internal regulatory resources.